MedeA VASP 6 Access the World’s Leading First-Principles DFT Code
At-a-Glance
MedeA®[1] VASP 6 provides industrial strength, efficient, cutting edge access to VASP, the world’s leading first-principles DFT code. Integrated in the MedeA atomistic simulation environment, with comprehensive structural databases, model building tools, and fully automated property modules, MedeA VASP 6 has an easy to use graphical user interface, access to automation and large-scale high-throughput capabilities, efficient property calculation, and interactive analysis.
Key Benefits
High accuracy, high performance first-principles methods and properties
Access to the latest DFT methods and developments (and beyond)
Efficient selection of parameters, thorough validation, and testing
Efficient management of all calculation parameters and data
Licensing model maximizes exploitation of computational resources
VASP 6 is the world’s leading first-principles solid state electronic structure program for solids, surfaces, and interfaces [2]. Possessing a comprehensive array of advanced features, including hybrid functionals [3] and metaGGAs, the ability to incorporate dispersion interactions, and comprehensive and validated self-consistent PAW potentials, MedeA VASP 6 provides access to state of the art first-principles simulation methods in a comprehensive and easy to use package. Advanced features include linear response calculations for properties such as Born effective charges, dielectric and piezoelectric tensors and NMR chemical shifts.
In terms of accuracy VASP 6 advances far beyond capabilities of semi-local and non-local functionals based on the unique, highly precise treatment of electronic correlation in the framework of the so-called ACFDT-RPA approach [4]. Outstanding results can be obtained even for systems where other ab initio approaches are known to fail. On the other hand, typical limitations of ab initio methods in terms of length and time scales are overcome by means of on-the-fly machine-learning of forcefields (MLFF) [5] during ab initio molecular dynamics, thus enabling unprecedented long dynamics runs on rather large systems. The obtained forcefields can be applied for many other simulations, such as structure optimizations, phonons, elastic properties etc..
MedeA VASP 6 is fully integrated in the MedeA Environment with graphical user interface driven model construction, efficient calculation execution, and analysis capabilities.
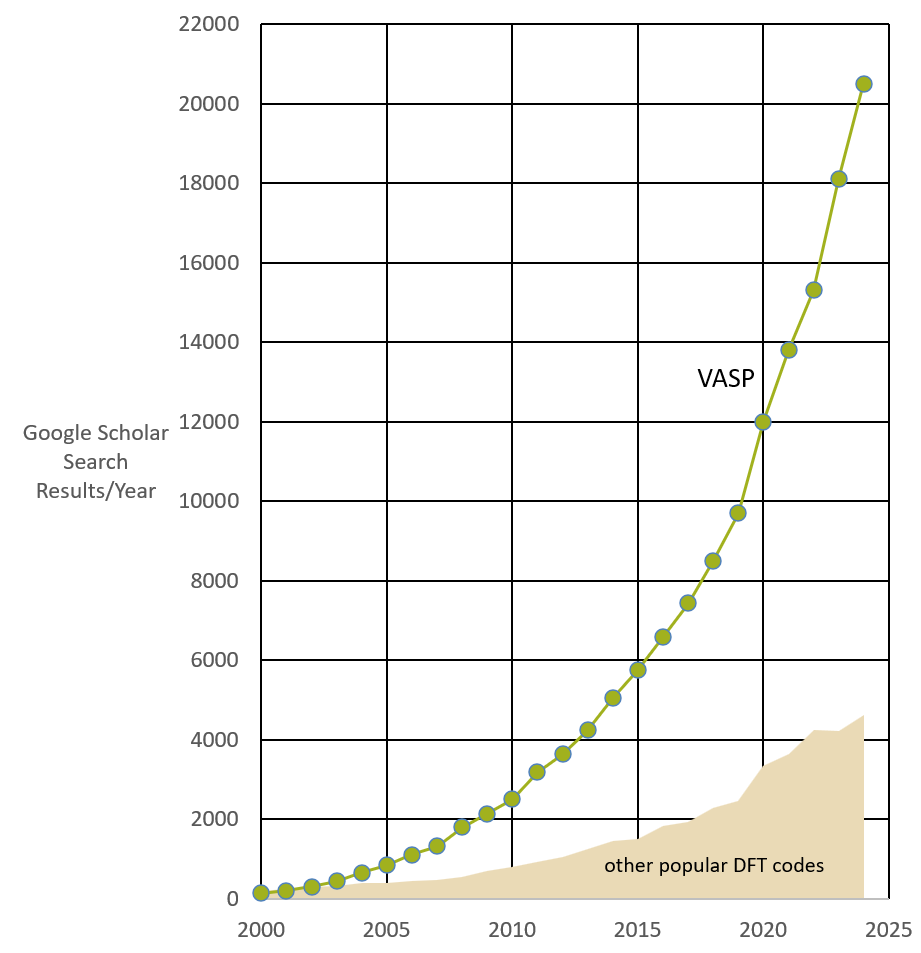
VASP is by far the most widely used ab-initio code applicable to solids and surfaces. This can easily be verified by an internet search.
Properties from MedeA VASP 6
MedeA VASP 6 enables the efficient computation of the following properties:
Total electronic energy of any 3D periodic arrangement of atoms, formation energy
Forces on atoms, pressure, and stress tensors
Collinear and non-collinear magnetic moments (optionally constrain total moment or atomic spin vectors)
Equilibrium lattice parameters and atomic positions as obtained from energy, force, and stress minimization, selective lattice parameter and atom position optimization
Ab-initio molecular dynamics: nVE, nVT, npT, npH ensembles, simulated annealing, lattice constraints, averages, uncertainties, and trajectories
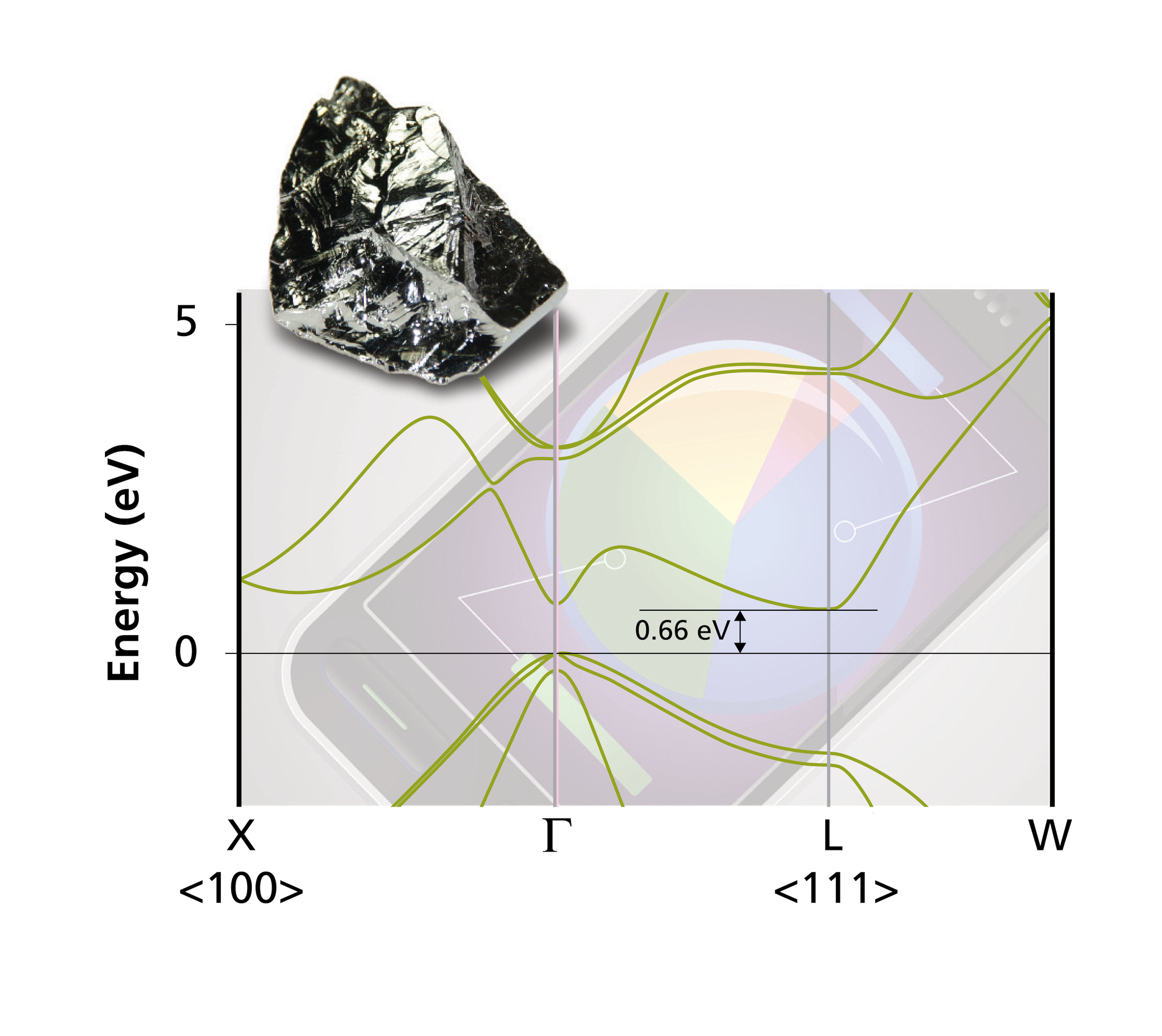
Energy band structure: atom and orbital momentum projected bands display (so-called fat bands), accurate band gaps, dopant levels, and band offsets based on hybrid functionals, and GW methods
Total and partial (atom, orbital momentum and magnetic quantum number projected) electronic density of states
Electronic charge and spin density, electrostatic potential, and Bader charge analysis
Work functions
Response functions including dielectric and piezoelectric tensors
Born effective charges and \(\Gamma\)-point phonon modes
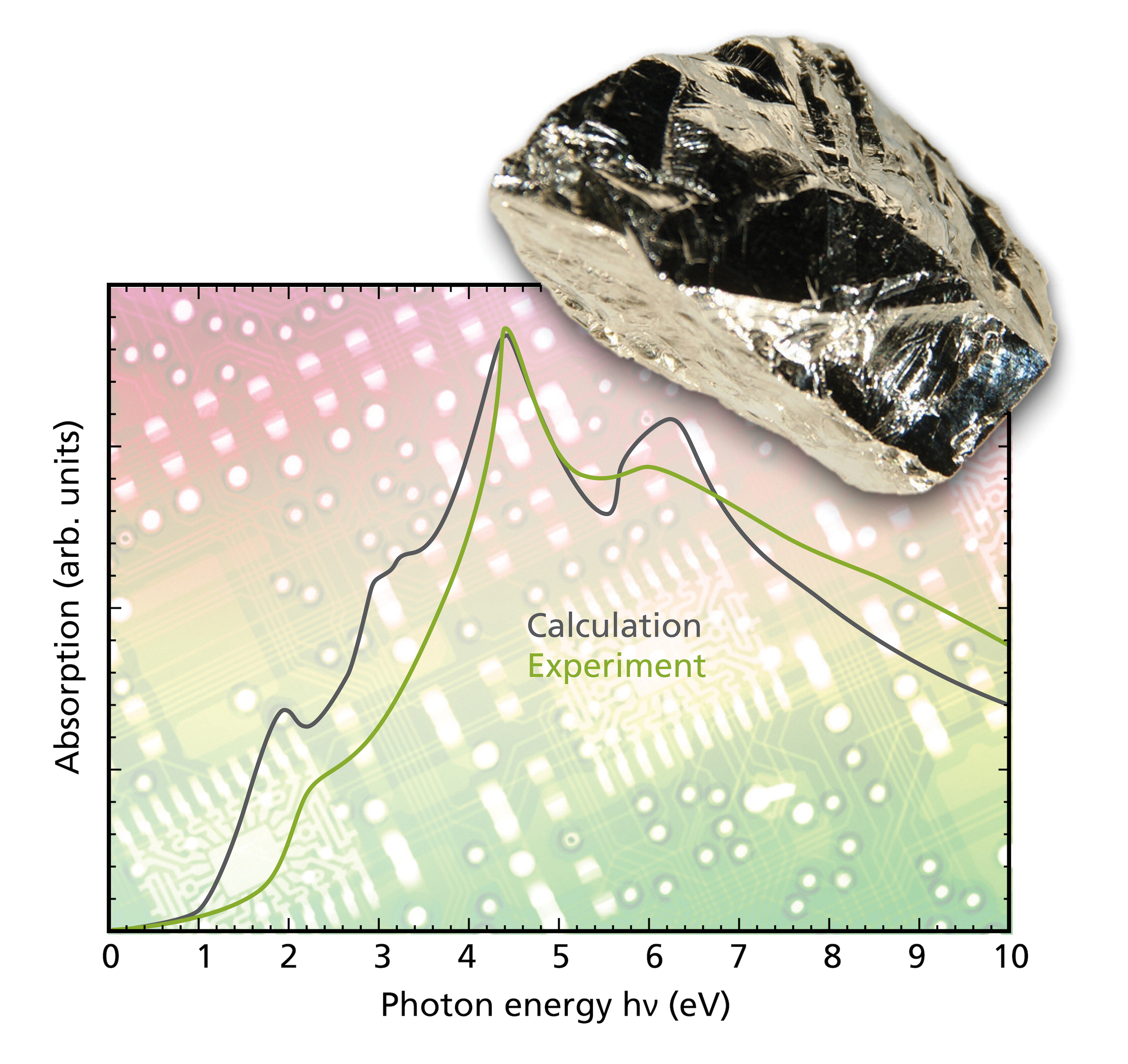
Optical spectra: dielectric function and conductivity, reflectivity, refractive index, transmission, absorption, attenuation and extinction coefficients as well as emissivity as a function of frequency, total emissivity vs. temperature, color spaces for D65 and FL2 spectral distributions of illuminants (CIELAB)
Hyperfine splitting
Electric field gradients and quadrupolar coupling constants
NMR chemical shifts
Solvation effects for surfaces and molecules
External electrostatic field effects
Computational Characteristics
Plane-wave based electronic structure method for periodic structures
All-electron method with projector augmented wave (PAW) potentials for all elements from H to Cm, including a set for highly accurate excited states
Scalar- and fully-relativistic, spin-orbit coupling
Density functional theory (DFT) with local (LDA) and gradient- corrected (GGA) semi-local functionals: AM05, PBEsol, PBE, revPBE, rPBE, BLYP, etc.
Hybrid functionals: HSE06, HSE03, PBE0, RSHXLDA, RSHXPBE, B3LYP, SCAN0, and dielectric-dependent hybrid functionals with the mixing parameter for non-local exchange being determined from the dielectric function. In addition, screened exchange and Hartree-Fock
Meta-GGA functionals: revTPSS, TPSS, SCAN, rSCAN, r 2SCAN, SCAN-L, rSCAN-L, r 2SCAN-L, v1-sregTM, v2-sregTM, v3-sregTM, v2-sregTM-L, OFR2, MS2, MS1, MS0, M06-L, modified Becke-Johnson LDA and its local variant
A variety of Van-der-Waals functionals: optB86b-vdW, optB88-vdW, optPBE-vdW, BEEF-vdW, rev-vdW-DF2, rPW86-vdW2, revPBE-vdW, vdW-DF-cx, rVV10, SCAN + rVV10, r 2SCAN + rVV10, PBE + rVV10L
DFT-D2/D3 (Grimme), DFT-dDsC, DFT-ulg, many-body dispersion energy, and Tkatchenko-Scheffler force-field based correction for van-der-Waals and dispersion forces and energies
Optical response functions from DFT, hybrid functionals, GW, or the time evolution algorithm
Electron-hole interactions (excitonic effects) from time-dependent hybrid functionals or solving the Bethe-Salpeter equation on top of GW [6]
Accurate total energy, forces, and zone center phonon modes from adiabatic connection fluctuation dissipation theorem and the random phase approximation (ACFDT-RPA), automatic optimization of atom positions based on ACFDT-RPA
Space-time algorithm for cumputing the polarizability for GW and ACFDT-RPA [7], which scales mostly cubic rather than quartic with system size, thus enabling simulations for much larger systems
Accurate total energy from Moeller-Plesset perturbation theory
Density functional perturbation theory, linear response
Electron-phonon coupling from stochastic displacement sampling (single configuration or Monte Carlo sampling)
On-the-fly machine-learning based on Bayesian error prediction during molecular dynamics simulations
Required Modules
MedeA Environment
MedeA VASP 6
Tightly Integrated Modules
MedeA Phonon
MedeA Electronics (Fermi surface and transport)
MedeA Mechanical Properties (MT)
MedeA Transition State Search
MedeA UNiversal CLuster Expansion (UNCLE)
MedeA Forcefield Optimizer
To accelerate calculations, MedeA VASP 6 uses intermediate results to improve the efficiency of subsequent steps. The MedeA JobServer and TaskServer architecture provides efficient storage and deployment for temporary files, and lets you focus on the science while computational bookkeeping and data storage is handled by the MedeA infrastructure. MedeA manages computational details, such as matching k-meshes and setting reasonable VASP parameters, automatically.
Tested, Validated, and Optimized for High-throughput
Materials Design supports a wide array of hardware configurations with optimized and validated VASP executables. Windows and Linux versions allow you to mix and match architectures, so you can run more calculations, and with larger models, on Linux clusters. You can be confident that the results will be consistent with calculations executed on your laptop or desktop machines. The MedeA Environment enables both investigative and high-throughput calculations, and the Materials Design licensing model allows you to maximize parallel execution. You can exploit the growing availability of high performance compute resources, and rapidly obtain state of the art research results. VASP executables supporting GPUs are available.
Find Out More
Watch the Webinar: VASP in MedeA - a fast way from models to reliable results and the online video tutorial: How to Calculate Elastic Constants with MedeA VASP. Learn how MedeA VASP can be employed by visiting the Materials Design Application Notes page of our website. Examples of MedeA VASP are in the following application notes:
Adsorption and Dissociation of Iodine Molecules on a Zr Surface
Acidity of Amorphous Silica-Alumina Catalysts
Stability of Alkaline-Earth Hydrides
Graphite Electrode Elastic Properties upon Li Intercalation
Atomic Structure of Hydrodesulfurization (HDS) Catalysts
Embrittlement of Cu Micro-Structures
Diffusion of Hydrogen in Nickel
Energy of Dissociative Chemisorption of SiH4 on Si (001) Surface
Structure of an iron oxide (Fe2O3) surface, as function of temperature and O2 pressure
Low-Strain Cathode Materials for Solid-State Li-Ion Batteries
Surface Magnetism of Fe(001)
Prediction of Schottky Barrier in Electronic Devices
Catalysts activity computational screening
Thermoelectric Properties of Bi2Te3 as calculated using MedeA Electronics
Accurate Band Gaps of Correlated Transition-Metal Oxides from Hybrid-Functional Calculations
Temperature-Dependent Phase Transitions of ZrO2
- download: