MedeA LAMMPS - A Powerful Gateway to a Powerful Simulation Program
✓ Part of the standard MedeA Environment
At-a-Glance
MedeA®[1] LAMMPS provides flexible calculation setup and analysis capabilities to unlock the power of LAMMPS.
LAMMPS [2] is one of the world’s leading forcefield-based molecular dynamics codes. Developed at Sandia National Laboratories by Steve Plimpton and fellow researchers, it enables highly efficient execution of forcefield based simulations in order to exploit large scale parallel and GPU-enabled computer architectures.
Key Benefits
Saves time and avoids mistakes by preparing complex input structure files in seconds rather than hours or days
Generates commands needed to run simulations automatically, without having to learn the language and syntax in detail
Identifies all required forcefield terms and assigns appropriate parameters according to the chosen forcefield
Analyzes key property output, and performs statistical analysis to determine averages and precision (error bars)
Specifications
Uses the LAMMPS forcefield engine for high performance on any computer, whether it be a scalar workstation or a massively parallel cluster
Runs on Windows (Server 2008/2012/2016, 7/8/10) and Linux (CentOS, Red Hat, Ubuntu, Debian, SUSE)
Supports 64-bit CPU architectures and computers with GPU cards [3]
MedeA LAMMPS calculations runs on an unlimited number of server computers or compute cores
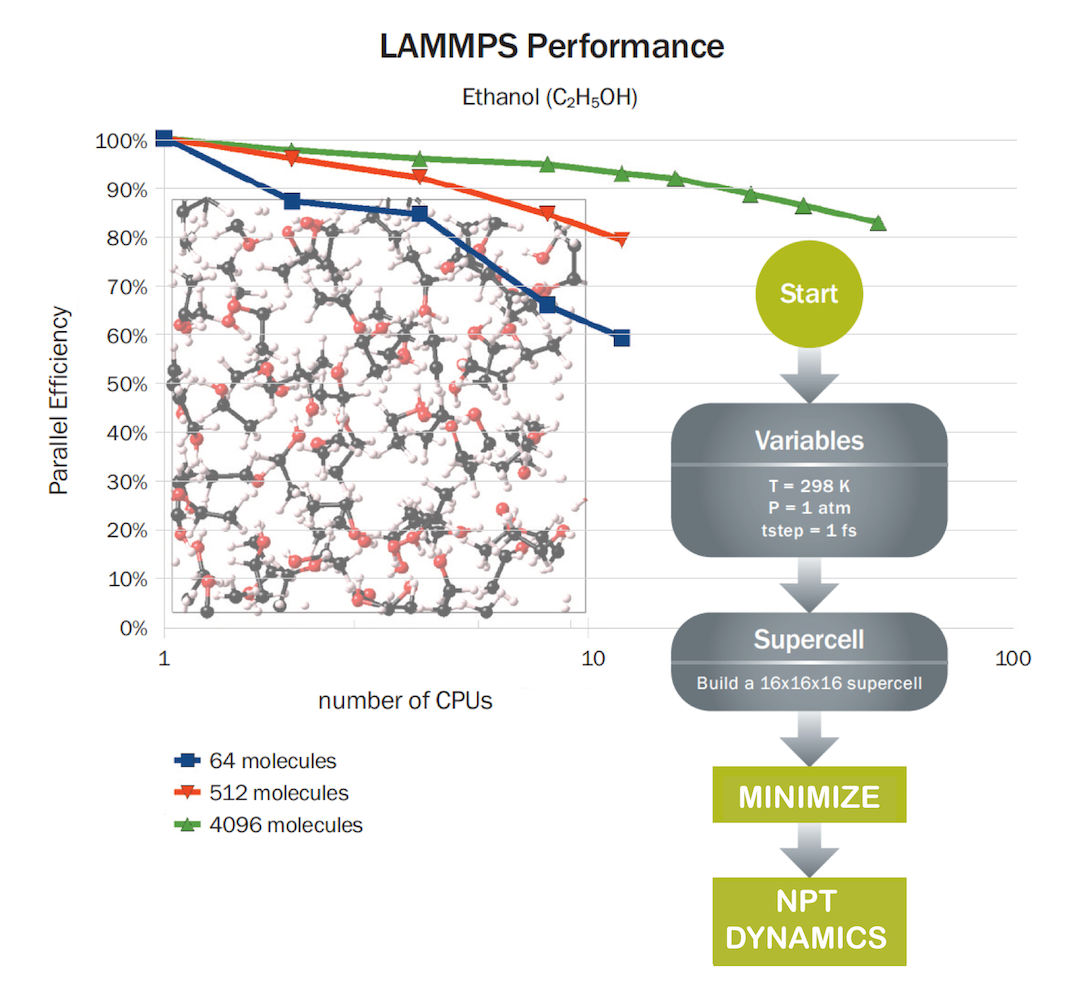
Setting up LAMMPS calculations on complex models using the flowchart interface is a significant productivity enhancer, and accumulating results in tables with the option to add occasional custom commands is very powerful indeed
Key Features
MedeA LAMMPS automates the details of properly formatting molecules, fluids, or solids into the required LAMMPS coordinate, connectivity, forcefield parameter, and command-line formats
Provides access to the core capabilities of LAMMPS:
Minimization
Molecular Dynamics simulations within the NVE, NVT, and NPT ensembles
Energy and Energy Derivative (force) calculation
Works with the MedeA JobServer and TaskServer to run your calculations on the appropriate, possibly distributed, hardware, while keeping the results well organized within the JobServer
Quick verification of all final and intermediate results through the convenient MedeA JobServer web browser interface
Full integration with MedeA Forcefield for advanced forcefield handling and assignment
Any custom forcefield which is provided in the appropriate MedeA Forcefield format is compatible
Powerful MedeA Flowcharts enable you to set up complex calculations with ease by graphically connecting stages
Flowcharts from any previous MedeA LAMMPS calculation can be re-used, edited, shared with colleagues, and rerun, even on different systems and compute servers
Provides options for expert LAMMPS users to add any LAMMPS commands to existing protocols, or to prepare completely customized simulations
Properties
After each calculation, MedeA LAMMPS automatically determines the block averages and fluctuations of:
Temperature
Pressure
Density
Cell parameters
Total energy and all components (potential, kinetic, Coulomb, and van der Waals)
Stress tensor elements
Visualization of trajectories of MD simulations and structure optimizations
Required Modules
MedeA Environment
MedeA LAMMPS (Part of the standard MedeA Environment)
Recommended Modules
MedeA HT-Launchpad
MedeA Amorphous Materials Builder
Supported Modules
MedeA CED
MedeA Diffusion
MedeA Surface Tension
MedeA Thermal Conductivity
MedeA Viscosity
MedeA Mechanical Properties (MT)
MedeA COMB3
Tightly Integrated Modules
MedeA Forcefield Optimizer
MedeA HT-Descriptors
MedeA Phonon
MedeA UNiversal CLuster Expansion (UNCLE)
Find Out More
Learn more about how MedeA LAMMPS can be used to study a broad range of inorganic, metallic, semiconductor, and organic materials by viewing the following webinar:
Harness the Power of LAMMPS Molecular Dynamics Code with MedeA
- download: